Tutorial¶
Note
Important! Before running any command, do not forget to change directory to vtam and activate the conda environment.
cd vtam
conda activate vtam
Note
With the exception of BLAST database files and the sqlite database all I/O files of VTAM are text files, that can be opened and edited by a simple text editor (gedit, geany, Notepad++ etc.):
- TSV: Text files with tab separated values. Can also be opened by spreadsheets such as LibreOffice, Excel
- YML: Text files used to provide parameter names and values
Data¶
In this tutorial, we use a small test dataset based from our previous publication: PMID 28776936. In this dataset, each sample was amplified by two overlapping markers (mfzr and zfzr), targeting the first 175-181 nucleotides of the COI gene (Fig. 2). We had three PCR replicates for each sample-marker combination (Fig. 1). The samples are tagged, so the combination of the forward and reverse tags can be used to identify the origin (sample) of each read.
To reduce run time, the test dataset contains only one mock sample, one negative control and two real samples.
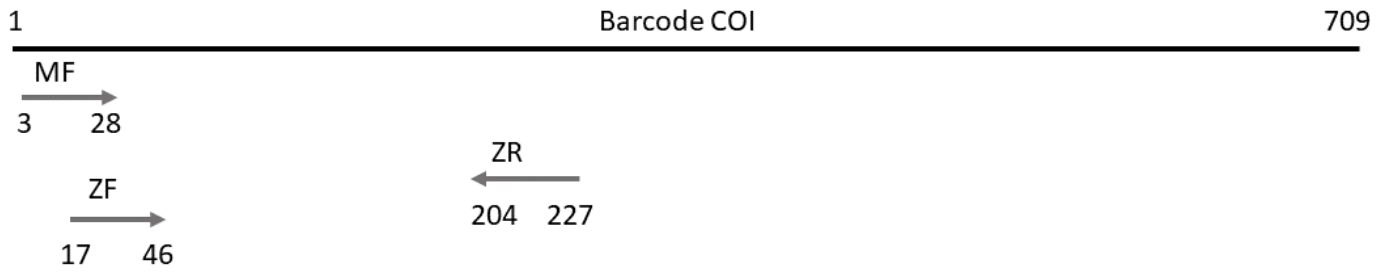
Figure 2. Positions on the primer on the COI gene used in the test dataset.
You can download these FASTQ files from here with this command:
wget -nc https://github.com/aitgon/vtam/releases/latest/download/fastq.tar.gz -O fastq.tar.gz
tar zxvf fastq.tar.gz
rm fastq.tar.gz
This will create a “FASTQ” directory with 12 FASTQ files:
$ ls fastq/
mfzr_1_fw.fastq
mfzr_1_rv.fastq
...
mfzr_1_fw.fastq: Forward reads of replicates of the MFZR marker (all samples)
merge: Merge FASTQ files¶
The simplest use of vtam is to analyze one sequencing run (run1) and one marker (MFZR).
The first step is to merge the FASTQ files and transform them into fasta files. It can be skipped, if you have single end reads, or your paired sequences have already been merged and transformed into fasta files.
Create a TSV (tab-separated file), with a header and 10 columns with all the information per FASTQ file pair.
We will call it fastqinfo_mfzr.tsv in this tutorial and you can download it here: fastqinfo_mfzr.tsv. This TSV file will determine, which file pairs should be merged. These files should be all in the fastq directory. This directory can contain other files as well, but they will not be analyzed.
The following columns are required in the fastqinfo_mfzr.tsv:
- TagFwd
- PrimerFwd
- TagRev
- PrimerRev
- Marker
- Sample
- Replicate
- Run
- FastqFwd
- FastqRev
Tag and primer sequences are in 5’ => 3’ orientation.
Hereafter are the first lines of the fastqinfo_mfzr.tsv file:
TagFwd PrimerFwd TagRev PrimerRev Marker Sample Replicate Run FastqFwd FastqRev
tcgatcacgatgt TCCACTAATCACAARGATATTGGTAC tgtcgatctacagc WACTAATCAATTWCCAAATCCTCC mfzr tpos1_run1 1 run1 mfzr_1_fw.fastq mfzr_1_rv.fastq
agatcgtactagct TCCACTAATCACAARGATATTGGTAC tgtcgatctacagc WACTAATCAATTWCCAAATCCTCC mfzr tnegtag_run1 1 run1 mfzr_1_fw.fastq mfzr_1_rv.fastq
We propose to work in a project directory called asper1 (the dataset comes from a project on Zingel asper) and copy user created input files such as fastqinfo_mfzr.tsv to the asper1/user_input directory.
asper1
`-- user_input
`-- fastqinfo_mfzr.tsv
fastq
|-- mfzr_1_fw.fastq
|-- mfzr_1_rv.fastq
|-- ...
Run merge for all file-pairs in the fastqinfo_mfzr.tsv
vtam merge --fastqinfo asper1/user_input/fastqinfo_mfzr.tsv --fastqdir fastq --fastainfo asper1/run1_mfzr/fastainfo.tsv --fastadir asper1/run1_mfzr/merged -v --log asper1/vtam.log
Note
For info on I/O files see the Reference section
This command adds a merged directory and a new fastainfo_mfzr.tsv file:
asper1
|-- run1_mfzr
| |-- fastainfo.tsv
| `-- merged
| |-- mfzr_1_fw.fasta
| |-- mfzr_2_fw.fasta
| `-- mfzr_3_fw.fasta
|-- user_input
| |-- fastqinfo_mfzr.tsv
|-- vtam.err
`-- vtam.log
fastq
|-- mfzr_1_fw.fastq
|-- mfzr_1_rv.fastq
|-- ...
The first lines of the fastainfo_mfzr.tsv look like this:
run marker sample replicate tagfwd primerfwd tagrev primerrev mergedfasta
run1 mfzr tpos1_run1 1 tcgatcacgatgt TCCACTAATCACAARGATATTGGTAC tgtcgatctacagc WACTAATCAATTWCCAAATCCTCC mfzr_1_fw.fasta
run1 mfzr tnegtag_run1 1 agatcgtactagct TCCACTAATCACAARGATATTGGTAC tgtcgatctacagc WACTAATCAATTWCCAAATCCTCC mfzr_1_fw.fasta
random_seq: Create a smaller randomized dataset from the main dataset (Optional)¶
The random_seq command is designed to create a smaller dataset with a given number of randomly selected sequences in each of its output files. This can be used to have the same number of reads for each replicates, and to reduce the number of reads. The input fasta files are listed in the fastainfo.tsv file.
vtam random_seq --fastainfo asper1/run1_mfzr/fastainfo.tsv --fastadir asper1/run1_mfzr/merged --random_seqdir asper1/run1_mfzr/randomized --random_seqinfo asper1/run1_mfzr/random_seq_info.tsv --samplesize 30000 -v
Note
For info on I/O files see the Reference section
The FASTA files with the randomized reads are written to the asper1/run1_mfzr/randomized directory:
asper1
|-- run1_mfzr
| |-- fastainfo.tsv
| |-- random_seq_info.tsv
| |--...
| `-- randomized
| |-- mfzr_1_fw_sampled.fasta
| |-- mfzr_2_fw_sampled.fasta
| |-- mfzr_3_fw_sampled.fasta
| `--
|-- ...
...
In addition, the TSV file asper1/run1_mfzr/random_seq_info.tsv is created which is the updated version of the fastainfo.tsv file. The random_seq_info.tsv file looks like this:
run marker sample replicate tagfwd primerfwd tagrev primerrev mergedfasta
run1 mfzr tpos1_run1 1 tcgatcacgatgt TCCACTAATCACAARGATATTGGTAC tgtcgatctacagc WACTAATCAATTWCCAAATCCTCC mfzr_1_fw_sampled.fasta
run1 mfzr tnegtag_run1 1 agatcgtactagct TCCACTAATCACAARGATATTGGTAC tgtcgatctacagc WACTAATCAATTWCCAAATCCTCC mfzr_1_fw_sampled.fasta
sortreads: Demultiplex and trim the reads¶
There is a single command sortreads to demultiplex the reads according to tags and to trim off tags and primers.
The sortreads command is designed to deal with a dual indexing, where forward and reverse tag combinations are used to determine the origin of the reads. This is one of the most complex case of demultiplexing, therefore we implemented sortreads to help users.
For simpler cases, we suggest using cutadapt directly, since it is quite straightforward.
In this example, we run the sortreads command on the output files of the random_seq, but if you have skipped the random_seq, it is possible to run it directly on the output of merge.
vtam sortreads --fastainfo asper1/run1_mfzr/random_seq_info.tsv --fastadir asper1/run1_mfzr/randomized --sorteddir asper1/run1_mfzr/sorted -v --log asper1/vtam.log
Note
For info on I/O files see the Reference section
The FASTA files with the sorted reads are written to the asper1/sorted directory:
asper1
|-- run1_mfzr
| |-- fastainfo.tsv
| |-- ...
| `-- sorted
| |-- run1_MFZR_14ben01_1_mfzr_1_fw_sampled_trimmed.fasta
│ |-- run1_MFZR_14ben01_2_mfzr_2_fw_sampled_trimmed.fasta
| |-- ...
| `-- sortedinfo.tsv
|-- ...
...
In addition, the TSV file asper1/run1_mfzr/sorted/sortedinfo.tsv lists the information, i.e. run, marker, sample and replicate about each sorted FASTA file. The sortedinfo.tsv file looks like this:
run marker sample replicate sortedfasta
run1 MFZR tpos1_run1 1 run1_MFZR_14ben01_1_mfzr_1_fw_sampled_trimmed.fasta
run1 MFZR tnegtag_run1 1 run1_MFZR_14ben01_2_mfzr_2_fw_sampled_trimmed.fasta
filter: Filter variants and create the ASV table¶
The filter command is typically first run with default parameters. From the output, users should identify clearly unwanted (‘delete’) and clearly necessary (‘keep’) occurrences (see Reference section for details). These false positive and false negative occurrences will be used as input to the optimize command. The optimize command will then suggest an optimal parameter combination tailored to your dataset. Then filter command should be run again with the optimized parameters.
Let’s run first the filter command with default parameters.
vtam filter --db asper1/db.sqlite --sortedinfo asper1/run1_mfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_mfzr/sorted --asvtable asper1/run1_mfzr/asvtable_default.tsv -v --log asper1/vtam.log
Note
For info on I/O files see the Reference section
This command creates two new files db.sqlite and asvtable_mfzr_default.tsv:
asper1
|-- db.sqlite
|-- run1_mfzr
| |-- asvtable_default.tsv
|-- ...
...
The database asper1/db.sqlite contains one table by filter, and in each table occurrences are marked as deleted (filter_delete = 1) or retained (filter_delete = 0). This database can be opened with a sqlite browser program (For example, https://sqlitebrowser.org / or https://sqlitestudio.pl).
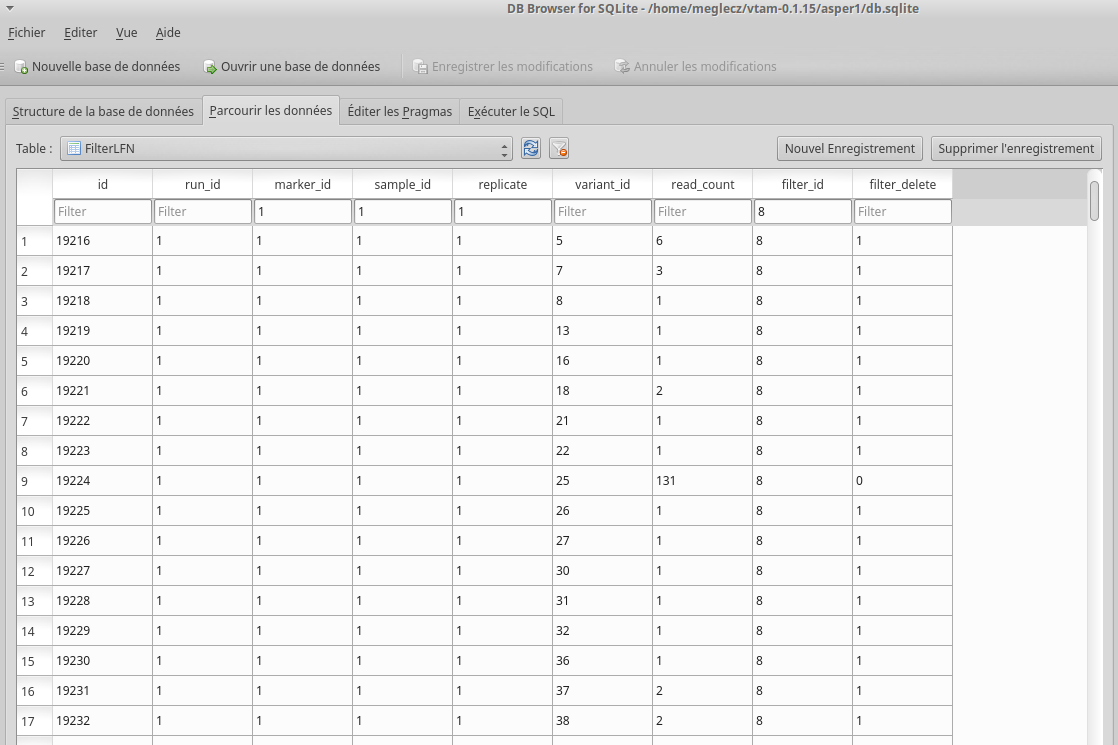
The asper1/run1_mfzr/asvtable_default.tsv contains information about the variants that passed all the filters such as the run, maker, read count over all replicates of a sample and the sequence. Hereafter are the first lines of the asvtable_default.tsv
run marker variant sequence_length read_count tpos1_run1 tnegtag_run1 14ben01 14ben02 clusterid clustersize chimera_borderline sequence
run1 MFZR 25 181 478 478 0 0 0 25 1 False ACTATACCTTATCTTCGCAGTATTCTCAGGAATGCTAGGAACTGCTTTTAGTGTTCTTATTCGAATGGAACTAACATCTCCAGGTGTACAATACCTACAGGGAAACCACCAACTTTACAATGTAATCATTACAGCTCACGCATTCCTAATGATCTTTTTCATGGTTATGCCAGGACTTGTT
run1 MFZR 51 181 165 0 0 0 165 51 1 False ACTATATTTAATTTTTGCTGCAATTTCTGGTGTAGCAGGAACTACGCTTTCATTGTTTATTAGAGCTACATTAGCGACACCAAATTCTGGTGTTTTAGATTATAATTACCATTTGTATAATGTTATAGTTACGGGTCATGCTTTTTTGATGATCTTTTTTTTAGTAATGCCTGCTTTATTG
Note
Filter can be run with the known_occurrences argument that will add an additional column for each mock sample flagging expected variants. This helps in creating the known_occurrences.tsv input file for the optimization step. For details see the Reference section
taxassign: Assign variants of ASV table to taxa¶
The taxassign command assigns ASV sequences in the last column of a TSV file such as the asvtable_default.tsv file to taxa.
The taxassign command needs a BLAST database (containing reference sequences of known taxonomic origin) and the taxonomy information file.
You can download the latest version of the COInr database formatted to VTAM from OSF (https://osf.io/vrfwz/). This database contains sequneces from Genbank and BOLD. If you want to customize it, you can use mkCOInr.
Download and unpack the database and the associated taxonomy file:
Note
You might need to update the URL and the file names for the latest version available in OSF
mkdir vtam_db
cd vtam_db
wget https://osf.io/9qyzf/download -O COInr_vtam_taxonomy_2022_05_06.tsv.gz
gunzip COInr_vtam_taxonomy_2022_05_06.tsv.gz
wget https://osf.io/qyr3b/download -O COInr_blast_2022_05_06.tar.gz
tar -zxvf COInr_blast_2022_05_06.tar.gz
rm COInr_blast_2022_05_06.tar.gz
These commands result in these new files:
vtam_db
├── COInr_blast_2022_05_06
│ ├── COInr_blast_2022_05_06.nhr
│ ├── COInr_blast_2022_05_06.nin
│ ├── COInr_blast_2022_05_06.nog
│ ├── COInr_blast_2022_05_06.nsd
│ ├── COInr_blast_2022_05_06.nsi
│ └── COInr_blast_2022_05_06.nsq
└── COInr_vtam_taxonomy_2022_05_06.tsv
Note
Alternatively, you can use your own custom database or the NCBI nucleotide database Reference section
Then, we can carry out the taxonomic assignation of variants in the asvtable_default.tsv with the following command:
vtam taxassign --db asper1/db.sqlite --asvtable asper1/run1_mfzr/asvtable_default.tsv --output asper1/run1_mfzr/asvtable_default_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 -v --log asper1/vtam.log
Note
For info on I/O files see the Reference section
This results in an additional file:
asper1/
|-- run1_mfzr
| |-- asvtable_default.tsv
| |-- asvtable_default_taxa.tsv
make_known_occurrences: Create file containing the known_occurences.tsv to be used as an inut for optimize¶
The make_known_occurrences command is designed to automatically create files containing the known and the missing occurences and it can be run if the expected occurrences in the mock samples are known in advance. Otherwise, it should be created by hand, based on the asvtable_default_taxa.tsv file.
The make_known_occurrences command requires the sample_types and mock_composition files as in input.
The exemple files can be downloaded from here: sample_types.tsv, mock_composition_mfzr.tsv
vtam make_known_occurrences --asvtable asper1/run1_mfzr/asvtable_default_taxa.tsv --sample_types asper1/user_input/sample_types.tsv --mock_composition asper1/user_input/mock_composition_mfzr.tsv --known_occurrences asper1/run1_mfzr/known_occurrences_mfzr.tsv --missing_occurrences asper1/run1_mfzr/missing_occurrences_mfzr.tsv -v
Note
For info on I/O files see the Reference section
The output is the known_occurrences_mfzr.tsv file wich can be used in the optimize step. If there are missing expected variants, they are written to the missing_occurrences_mfzr.tsv file.
optimize: Compute optimal filter parameters based on mock and negative samples¶
The optimize command helps users choose optimal parameters for filtering that are specifically adjusted to the dataset. This optimization is based on mock samples and negative controls.
Users should prepare a TSV file (known_occurrences_mfzr.tsv) with occurrences to be kept in the results (typically expected variants of the mock samples) and occurrences to be clearly deleted (typically all occurrences in negative controls, and unexpected occurrences in the mock samples). For details see the Reference section.
The example TSV file for the known occurrences of the MFZR marker can be found here : known_occurrences_mfzr.tsv.
The first lines of this file look like this:
marker run sample mock variant action sequence
MFZR run1 tpos1_run1 1 keep ACTATATTTTATTTTTGGGGCTTGATCCGGAATGCTGGGCACCTCTCTAAGCCTTCTAATTCGTGCCGAGCTGGGGCACCCGGGTTCTTTAATTGGCGACGATCAAATTTACAATGTAATCGTCACAGCCCATGCTTTTATTATGATTTTTTTCATGGTTATGCCTATTATAATC
MFZR run1 tpos1_run1 1 keep ACTTTATTTTATTTTTGGTGCTTGATCAGGAATAGTAGGAACTTCTTTAAGAATTCTAATTCGAGCTGAATTAGGTCATGCCGGTTCATTAATTGGAGATGATCAAATTTATAATGTAATTGTAACTGCTCATGCTTTTGTAATAATTTTCTTTATAGTTATACCTATTTTAATT
...
MFZR run1 tpos1_run1 1 delete TTTATATTTCATTTTTGGTGCATGATCAGGTATGGTGGGTACTTCCCTTAGTTTATTAATTCGAGCAGAACTTGGTAATCCTGGTTCTTTGATTGGCGATGATCAGATTTATAACGTTATTGTCACTGCCCATGCTTTTATTATGATTTTTTTTATAGTGATACCTATTATAATT
MFZR run1 tnegtag_run1 0 delete TTTATATTTTATTTTTGGAGCCTGAGCTGGAATAGTAGGTACTTCCCTTAGTATACTTATTCGAGCCGAATTAGGACACCCAGGCTCTCTAATTGGAGACGACCAAATTTATAATGTAATTGTTACTGCTCATGCTTTTGTAATAATTTTTTTTATAGTTATGCCAATTATAATT
Note
It is possible to add extra columns with your notes (for example taxon names) to this file after the sequence column. They will be ignored by VTAM.
The optimize command is run like this:
vtam optimize --db asper1/db.sqlite --sortedinfo asper1/run1_mfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_mfzr/sorted --known_occurrences asper1/run1_mfzr/known_occurrences_mfzr.tsv --outdir asper1/run1_mfzr -v --log asper1/vtam.log
Note
For info on I/O files see the Reference section
This command creates four new files:
asper1/
|-- db.sqlite
|-- run1_mfzr
| |-- ...
| |-- optimize_lfn_sample_replicate.tsv
| |-- optimize_lfn_read_count_and_lfn_variant.tsv
| |-- optimize_lfn_variant_specific.tsv
| |-- optimize_pcr_error.tsv
Note
Running vtam optimize will run three underlying scripts:
- OptimizePCRerror, to optimize pcr_error_var_prop
- OptimizeLFNsampleReplicate, to optimize lfn_sample_replicate_cutoff
- OptimizeLFNreadCountAndLFNvariant, to optimize lfn_read_count_cutoff and lfn_variant_cutoff.
While OptimizePCRerror and OptimizeLFNsampleReplicate do not depend on the other two parameters to be optimized, OptimizeLFNreadCountAndLFNvariant does. For a finer tuning, it is possible to run the three subscripts one by one, and use the optimized values of pcr_error_var_prop and lfn_sample_replicate_cutoff instead of their default values, when running OptimizeLFNreadCountAndLFNvariant. This procedure can propose less stringent values for lfn_read_count_cutoff and lfn_variant_cutoff, but still eliminate as many as possible unexpected occurrences, and keep all expected ones.
To run just one subscript, the –until flag can be added to the vtam optimize command
- until OptimizePCRerror
- unlit OptimizeLFNsampleReplicate
- until OptimizeLFNreadCountAndLFNvariant
e.g.
vtam optimize --db asper1/db.sqlite --sortedinfo asper1/run1_mfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_mfzr/sorted --known_occurrences asper1/run1_mfzr/known_occurrences_mfzr.tsv --outdir asper1/run1_mfzr -v --log asper1/vtam.log --until OptimizePCRerror
vtam optimize --db asper1/db.sqlite --sortedinfo asper1/run1_mfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_mfzr/sorted --known_occurrences asper1/run1_mfzr/known_occurrences_mfzr.tsv --outdir asper1/run1_mfzr -v --log asper1/vtam.log --until OptimizeLFNsampleReplicate
Based on the output, create a params_optimize_mfzr.yml file that will contain the optimal values suggested for lfn_sample_replicate_cutoff and pcr_error_var_prop
lfn_sample_replicate_cutoff: 0.003
pcr_error_var_prop: 0.1
Run OptimizeLFNreadCountAndLFNvariant with the optimized parameters for the above two parameters.
vtam optimize --db asper1/db.sqlite --sortedinfo asper1/run1_mfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_mfzr/sorted --known_occurrences asper1/run1_mfzr/known_occurrences_mfzr.tsv --outdir asper1/run1_mfzr -v --log asper1/vtam.log --until OptimizeLFNreadCountAndLFNvariant --params asper1/user_input/params_optimize_mfzr.yml
This step will suggest the following parameter values
lfn_variant_cutoff: 0.001
lfn_read_count_cutoff: 20
For simplicity, we continue the tutorial with parameters optimized previously, with running all 3 optimize steps in one command.
filter: Create an ASV table with optimal parameters and assign variants to taxa¶
See the Reference section on how to establish the optimal parameters from the outout of optimize. Once the optimal filtering parameters are chosen, rerun the filter command using the existing db.sqlite database that already has all the variant counts.
Make a params_mfzr.yml file that contains the parameter names and values that differ from the default settings.
The params_mfzr.yml can be found here: params_mfzr.yml and it looks like this:
lfn_variant_cutoff: 0.001
lfn_sample_replicate_cutoff: 0.003
lfn_read_count_cutoff: 70
pcr_error_var_prop: 0.1
Run filter with optimized parameters:
vtam filter --db asper1/db.sqlite --sortedinfo asper1/run1_mfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_mfzr/sorted --params asper1/user_input/params_mfzr.yml --asvtable asper1/run1_mfzr/asvtable_optimized.tsv -v --log asper1/vtam.log
Running again taxassign will complete the asvtable_optimized.tsv with the taxonomic information. It will be very quick since most variants in the table have already gone through the taxonomic assignment, and these assignations are extracted from the db.sqlite.
vtam taxassign --db asper1/db.sqlite --asvtable asper1/run1_mfzr/asvtable_optimized.tsv --output asper1/run1_mfzr/asvtable_optimized_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 -v --log asper1/vtam.log
We finished our first analysis with VTAM! The resulting directory structure looks like this:
asper1/
|-- db.sqlite
|-- run1_mfzr
| |-- asvtable_default.tsv
| |-- asvtable_default_taxa.tsv
| |-- asvtable_optimized.tsv
| |-- asvtable_optimized_taxa.tsv
| |-- fastainfo.tsv
| |-- merged
| | |-- mfzr_1_fw.fasta
| | |-- ...
| |-- optimize_lfn_sample_replicate.tsv
| |-- optimize_lfn_read_count_and_lfn_variant.tsv
| |-- optimize_lfn_variant_specific.tsv
| |-- optimize_pcr_error.tsv
| `-- sorted
| |-- run1_MFZR_14ben01_2_mfzr_2_fw_trimmed.fasta
| |-- ...
| `-- sortedinfo.tsv
Add new run-marker data to the existing database¶
The same samples can be amplified by different but strongly overlapping markers. In this case, it makes sense to pool all the data into the same database, and produce just one ASV table, with information of both markers. This is the case in our test dataset.
It is also frequent to have different sequencing runs (with one or several markers) that are part of the same study. Feeding them to the same database ensures coherence in variant IDs, and gives the possibility to easily produce one ASV table with all the runs and avoids re-running the taxassign on variants that have already been assigned to a taxon.
We assume that you have gone through the basic pipeline in the previous section.
Let’s see an example on how to complete the previous analyses with the dataset obtained for the same samples but for
another marker (ZFZR). The principle is the same if you want to complete the analyses with data from a different
sequencing run.
First, we need to prepare these user inputs:
The directory with the FASTQ files: fastqinfo_zfzr.tsv.
This is the merge command for the new run-marker:
vtam merge --fastqinfo asper1/user_input/fastqinfo_zfzr.tsv --fastqdir fastq --fastainfo asper1/run1_zfzr/fastainfo.tsv --fastadir asper1/run1_zfzr/merged -v --log asper1/vtam.log
This is the sortreads command for the new marker ZFZR:
vtam sortreads --fastainfo asper1/run1_zfzr/fastainfo.tsv --fastadir asper1/run1_zfzr/merged --sorteddir asper1/run1_zfzr/sorted -v --log asper1/vtam.log
The filter command for the new marker ZFZR is the same as in the basic pipeline, but we will complete the previous database asper1/db.sqlite with the new variants.
vtam filter --db asper1/db.sqlite --sortedinfo asper1/run1_zfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_zfzr/sorted --asvtable asper1/run1_zfzr/asvtable_default.tsv -v --log asper1/vtam.log
Next we run the taxassign command for the new ASV table asper1/asvtable_zfzr_default.tsv:
vtam taxassign --db asper1/db.sqlite --asvtable asper1/run1_zfzr/asvtable_default.tsv --output asper1/run1_zfzr/asvtable_default_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 -v --log asper1/vtam.log
Here, we prepare a new file of known occurrences for the ZFZR marker: known_occurrences_zfzr.tsv.
Then we run the optimize command with the known occurrences:
vtam optimize --db asper1/db.sqlite --sortedinfo asper1/run1_zfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_zfzr/sorted --known_occurrences asper1/user_input/known_occurrences_zfzr.tsv --outdir asper1/run1_zfzr -v --log asper1/vtam.log
At this point, we prepare a new params file for the ZFZR marker: params_zfzr.yml.
Then we run the filter command with the optimized parameters:
vtam filter --db asper1/db.sqlite --sortedinfo asper1/run1_zfzr/sorted/sortedinfo.tsv --sorteddir asper1/run1_zfzr/sorted --params asper1/user_input/params_zfzr.yml --asvtable asper1/run1_zfzr/asvtable_optimized.tsv -v --log asper1/vtam.log
Then we run the taxassign command of the optimized ASV table:
vtam taxassign --db asper1/db.sqlite --asvtable asper1/run1_zfzr/asvtable_optimized.tsv --output asper1/run1_zfzr/asvtable_optimized_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 -v --log asper1/vtam.log
At this point, we have run the equivalent of the previous section (MFZR marker) for the ZFZR marker.
Now we can pool the two markers MFZR and ZFZR. This input TSV file asper1/user_input/pool_run_marker.tsv defines the run and marker combinations that must be pooled. The pool_run_marker.tsv that looks like this:
run marker
run1 MFZR
run1 ZFZR
Then the pool command can be used:
vtam pool --db asper1/db.sqlite --runmarker asper1/user_input/pool_run_marker.tsv --asvtable asper1/asvtable_pooled_mfzr_zfzr.tsv --log asper1/vtam.log -v
Note
For info on I/O files see the Reference section.
The output asvtable_pooled_mfzr_zfzr.tsv is an asv table that contains all samples of all runs (in this example there is only one run), and all “unique” variants: variants identical in their overlapping regions are pooled into the one line.
Summing read count from different markers does not make sense. In asvtable_pooled_mfzr_zfzr.tsv cells contain 1/0 for presence/absence instead of read counts.
Hereafter are the first lines of the asvtable_pooled_mfzr_zfzr.tsv.
variant_id pooled_variants run marker tpos1_run1 tnegtag_run1 14ben01 14ben02 clusterid clustersize pooled_sequences sequence
25 25 run1 MFZR 1 0 0 0 25 1 ACTATACCTTATCTTCGCAGTATTCTCAGGAATGCTAGGAACTGCTTTTAGTGTTCTTATTCGAATGGAACTAACATCTCCAGGTGTACAATACCTACAGGGAAACCACCAACTTTACAATGTAATCATTACAGCTCACGCATTCCTAATGATCTTTTTCATGGTTATGCCAGGACTTGTT ACTATACCTTATCTTCGCAGTATTCTCAGGAATGCTAGGAACTGCTTTTAGTGTTCTTATTCGAATGGAACTAACATCTCCAGGTGTACAATACCTACAGGGAAACCACCAACTTTACAATGTAATCATTACAGCTCACGCATTCCTAATGATCTTTTTCATGGTTATGCCAGGACTTGTT
137 137 run1 MFZR 1 0 0 0 137 1 ACTTTATTTCATTTTCGGAACATTTGCAGGAGTTGTAGGAACTTTACTTTCATTATTTATTCGTCTTGAATTAGCTTATCCAGGAAATCAATTTTTTTTAGGAAATCACCAACTTTATAATGTGGTTGTGACAGCACATGCTTTTATCATGATTTTTTTCATGGTTATGCCGATTTTAATC ACTTTATTTCATTTTCGGAACATTTGCAGGAGTTGTAGGAACTTTACTTTCATTATTTATTCGTCTTGAATTAGCTTATCCAGGAAATCAATTTTTTTTAGGAAATCACCAACTTTATAATGTGGTTGTGACAGCACATGCTTTTATCATGATTTTTTTCATGGTTATGCCGATTTTAATC
1112 1112,4876 run1 MFZR,ZFZR 1 0 0 0 1112 1 CTTATATTTTATTTTTGGTGCTTGATCAGGGATAGTGGGAACTTCTTTAAGAATTCTTATTCGAGCTGAACTTGGTCATGCGGGATCTTTAATCGGAGACGATCAAATTTACAATGTAATTGTTACTGCACACGCCTTTGTAATAATTTTTTTTATAGTTATACCTATTTTAATT,TGCTTGATCAGGGATAGTGGGAACTTCTTTAAGAATTCTTATTCGAGCTGAACTTGGTCATGCGGGATCTTTAATCGGAGACGATCAAATTTACAATGTAATTGTTACTGCACACGCCTTTGTAATAATTTTTTTTATAGTTATACCTATTTTAATT CTTATATTTTATTTTTGGTGCTTGATCAGGGATAGTGGGAACTTCTTTAAGAATTCTTATTCGAGCTGAACTTGGTCATGCGGGATCTTTAATCGGAGACGATCAAATTTACAATGTAATTGTTACTGCACACGCCTTTGTAATAATTTTTTTTATAGTTATACCTATTTTAATT
The sequence column is a representative sequence of the pooled variants. pooled_sequeces is a list of pooled variants. For details see the ASV table format.
Complete the asvtable_pooled_mfzr_zfzr.tsv with taxonomic assignments using the taxassign command:
vtam taxassign --db asper1/db.sqlite --asvtable asper1/asvtable_pooled_mfzr_zfzr.tsv --output asper1/asvtable_pooled_mfzr_zfzr_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 --log asper1/vtam.log -v
We finished running VTAM for a second marker ZFZR.
The additional data for the ZFZR and the pooled data can be found here:
asper1
|-- asvtable_pooled_mfzr_zfzr.tsv
|-- asvtable_pooled_mfzr_zfzr_taxa.tsv
|-- ...
|-- run1_zfzr
| |-- asvtable_default.tsv
| |-- asvtable_default_taxa.tsv
| |-- asvtable_optimized.tsv
| |-- asvtable_optimized_taxa.tsv
| |-- ...
...
Running VTAM for data with several run-marker combinations¶
The outcome of some of the filtering steps (LFNfilter, renkonen) in vtam depends on the composition of the other samples in the dataset. Therefore vtam is designed to optimize parameters separately for each run-marker combination and do the filtering steps separately for each of them. However, since run-marker information is taken into account in the vtam scripts, technically it is possible to analyze several dataset (run-marker combination) in a single command.
Let’s use the same dataset as before, but run the two markers together. These analyses will give the same results as the previously described pipeline, we will just use fewer commands.
We will define a new output folder asper2 to clearly separate the results from the previous ones.
For the merge command, the format of fastqinfo file is as before, but it includes info on both markers
vtam merge --fastqinfo asper2/user_input/fastqinfo.tsv --fastqdir fastq --fastainfo asper2/run1/fastainfo.tsv --fastadir asper2/run1/merged -v --log asper2/vtam.log
These are the sortreads and the filter commands:
vtam sortreads --fastainfo asper2/run1/fastainfo.tsv --fastadir asper2/run1/merged --sorteddir asper2/run1/sorted -v --log asper2/vtam.log
vtam filter --db asper2/db.sqlite --sortedinfo asper2/run1/sorted/sortedinfo.tsv --sorteddir asper2/run1/sorted --asvtable asper2/run1/asvtable_default.tsv -v --log asper2/vtam.log
The asvtable_default.tsv file contains all variants that passed the filters from both markers. Variants identical in the overlapping regions are NOT pooled at this point, since the optimization will be done separately for each marker-run combination.
This is the taxassign command:
vtam taxassign --db asper2/db.sqlite --asvtable asper2/run1/asvtable_default.tsv --output asper2/run1/asvtable_default_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 -v --log asper2/vtam.log
For the optimize command, make one single known_occurrences.tsv file with known occurrences for both markers:
vtam optimize --db asper2/db.sqlite --sortedinfo asper2/run1/sorted/sortedinfo.tsv --sorteddir asper2/run1/sorted --known_occurrences asper2/user_input/known_occurrences.tsv --outdir asper2/run1 -v --log asper2/vtam.log
Each of the output files in the asper2/run1/optimize folder will contain information on both markers. The analyses suggest the optimal parameters have been run independently for the two markers. You have to choose the optimal parameters and make params.yml files separately for each of them.
Since the optimal parameters for the two markers are likely to be different, you have to run this step separately for the two markers.
The content of the sortedinfo is used to define the dataset for which the filtering is done. The asper2/sorted/sortedinfo.tsv contains information on both markers. That is why, the filtering by default parameters were run on both markers. In this step, you have to split this file in two. Each of them will contain info on only one marker.
So you will need the following input files:
- asper2/user_input/params_mfzr.yml
- asper2/user_input/params_zfzr.yml
- asper2/user_input/readinfo_mfzr.tsv
- asper2/user_input/readinfo_zfzr.tsv
Run filter for MFZR:
vtam filter --db asper2/db.sqlite --sortedinfo asper2/user_input/sortedinfo_mfzr.tsv --sorteddir asper2/run1/sorted --asvtable asper2/run1/asvtable_optimized_mfzr.tsv -v --log asper2/vtam.log --params asper2/user_input/params_mfzr.yml
Run filter for ZFZR:
vtam filter --db asper2/db.sqlite --sortedinfo asper2/user_input/sortedinfo_zfzr.tsv --sorteddir asper2/run1/sorted --asvtable asper2/run1/asvtable_optimized_zfzr.tsv -v --log asper2/vtam.log --params asper2/user_input/params_zfzr.yml
To end this case, we run the pool and taxassign commands:
vtam pool --db asper2/db.sqlite --runmarker asper2/user_input/pool_run_marker.tsv --asvtable asper2/pooled_asvtable_mfzr_zfzr.tsv --log asper2/vtam.log -v
vtam taxassign --db asper2/db.sqlite --asvtable asper2/pooled_asvtable_mfzr_zfzr.tsv --output asper2/pooled_asvtable_mfzr_zfzr_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 --log asper2/vtam.log -v
We finished running VTAM for the two markers!
Run VTAM with snakemake¶
Snakemake is a tool to create analysis workflows composed of several steps of VTAM. In this section, we will use a Snakefile to run several steps together.
This part of the tutorial supposes that you have read the tutorial on how to run vtam command by command for one run-marker combination and you understand the role of each step and the essential input files.
Basic pipeline with snakemake: one run-marker combination¶
We will work marker by marker. At the root there is a project folder (asper1). The analyses related to run-marker will go to different subfolders (eg. run1_mfzr) that will contain all related files. We will illustrate the pipeline with the marker MFZR but the same commands can be run later in the same project folder with the marker ZFZR. First make sure that you have the vtam_db and fastq directories as in the Data section of the Tutorial. To setup the pipeline we need the fastqinfo_mfzr.tsv file as before and a config file for snakemake, called snakeconfig_mfzr.yml. We will prepare these files inside a <project>/user_input folder as before. The snakeconfig_mfzr.yml looks like this:
project: 'asper1'
subproject: 'run1_mfzr'
fastqinfo: 'asper1/user_input/fastqinfo_mfzr.tsv'
fastqdir: 'fastq'
known_occurrences: 'asper1/user_input/known_occurrences_mfzr.tsv'
params: 'asper1/user_input/params_mfzr.yml'
blastdbdir: 'vtam_db/COInr_blast_2022_05_06'
blastdbname: 'COInr_blast_2022_05_06'
taxonomy: 'vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv'
Make sure the snakemake.yml is in the current working directory. The resulting file tree looks like this:
.
|-- asper1
| `-- user_input
| |-- fastqinfo_mfzr.tsv
| `-- snakeconfig_mfzr.yml
|-- fastq
| |-- mfzr_1_fw.fastq
| |-- ...
|-- snakefile.yml
`-- vtam_db
|-- COInr_blast_2022_05_06
| |-- COInr_blast_2022_05_06.nhr
| |-- ...
`-- COInr_vtam_taxonomy_2022_05_06.tsv
Steps merge, sortreads, filter with default parameters, taxassign¶
You can run these four steps in one go and create the ASV table with taxonomic assignments with this command:
snakemake --printshellcmds --resources db=1 --snakefile snakefile.yml --cores 4 --configfile asper1/user_input/snakeconfig_mfzr.yml --until asvtable_taxa
We find the same directory tree as before:
asper1
|-- db.sqlite
|-- run1_mfzr
| |-- asvtable.tsv
| |-- asvtable_taxa.tsv
| |-- fastainfo.tsv
| |-- merged
| | |-- mfzr_1_fw.fasta
| | |-- ...
| `-- sorted
| |-- run1_MFZR_14ben01_2_mfzr_2_fw_trimmed.fasta
| |-- ...
| `-- sortedinfo.tsv
|-- user_input
| |-- fastqinfo_mfzr.tsv
| |-- known_occurrences_mfzr.tsv
| |-- params_mfzr.yml
| `-- snakeconfig_mfzr.yml
|-- vtam.err
`-- vtam.log
The step optimize¶
You can now create the asper1/user_input/known_occurrences_mfzr.tsv based on the informations given by the asper1/run1_mfzr/asvtable_default_taxa.tsv.
Then you will run the optimize script to look for better parameters for the MFZR marker:
snakemake --printshellcmds --resources db=1 --snakefile snakefile.yml --cores 4 --configfile asper1/user_input/snakeconfig_mfzr.yml --until optimize
The resulting optimization files will be found here:
asper1
|-- db.sqlite
|-- run1_mfzr
| |-- ...
| |-- optimize_lfn_sample_replicate.tsv
| |-- optimize_lfn_read_count_and_lfn_variant.tsv
| |-- optimize_lfn_variant_specific.tsv
| |-- optimize_pcr_error.tsv
The steps filter with optimized parameters and taxassign¶
Define the optimal parameters and create a parameter file: asper1/user_input/params_mfzr.yml. Run the filtering and taxassign steps:
snakemake --printshellcmds --resources db=1 --snakefile snakefile.yml --cores 4 --configfile asper1/user_input/snakeconfig_mfzr.yml --until asvtable_optimized_taxa
This last command will give you two new ASV tables with optimized parameters:
asper1
|-- ...
|-- run1_mfzr
| |-- ...
| |-- asvtable_params_taxa.tsv
| |-- asvtable_params.tsv
Add new run-marker data to existing database¶
The same commands can be run for the second marker ZFZR. You will need the following additional files:
- asper1/user_input/snakeconfig_zfzr.yml
- asper1/user_input/fastqinfo_zfzr.tsv
- asper1/user_input/known_occurrences_zfzr.tsv
- asper1/user_input/params_zfzr.yml
The snakeconfig_zfzr.yml will look like this:
project: 'asper1'
subproject: 'run1_zfzr'
fastqinfo: 'asper1/user_input/fastqinfo_zfzr.tsv'
fastqdir: 'fastq'
known_occurrences: 'asper1/user_input/known_occurrences_zfzr.tsv'
params: 'asper1/user_input/params_zfzr.yml'
blastdbdir: 'vtam_db/COInr_blast_2022_05_06'
blastdbname: 'COInr_blast_2022_05_06'
taxonomy: 'vtam_db/COInr_vtam_taxonomy_2022_05_06'
Then you can run the same commands as above for the new marker ZFZR:
snakemake --printshellcmds --resources db=1 --snakefile snakefile.yml --cores 4 --configfile asper1/user_input/snakeconfig_zfzr.yml --until asvtable_taxa
snakemake --printshellcmds --resources db=1 --snakefile snakefile.yml --cores 4 --configfile asper1/user_input/snakeconfig_zfzr.yml --until optimize
snakemake --printshellcmds --resources db=1 --snakefile snakefile.yml --cores 4 --configfile asper1/user_input/snakeconfig_zfzr.yml --until asvtable_optimized_taxa
These commands will generate a new folder with the same files for the new marker ZFZR. The database db.sqlite will be shared by both markers MFZR and ZFZR:
asper1
|-- db.sqlite
|-- ...
|-- run1_zfzr
| |-- asvtable.tsv
| |-- asvtable_taxa.tsv
| |-- fastainfo.tsv
| |-- merged
| | |-- zfzr_1_fw.fasta
| | |-- ...
| `-- sorted
| |-- sortedinfo.tsv
| |-- run1_ZFZR_14ben01_2_mfzr_2_fw_trimmed.fasta
| |-- ...
The results of the two markers can be pooled as before:
vtam pool --db asper1/db.sqlite --runmarker asper1/user_input/pool_run_marker.tsv --asvtable asper1/pooled_asvtable_mfzr_zfzr.tsv --log asper1/vtam.log -v
vtam taxassign --db asper1/db.sqlite --asvtable asper1/pooled_asvtable_mfzr_zfzr.tsv --output asper1/pooled_asvtable_mfzr_zfzr_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 --log asper1/vtam.log -v
Running snakemake for data with several run-marker combinations¶
Similarly as before, we can run all run-markers simultaneously. We will use these files:
- asper2/user_input/snakeconfig.yml
- asper2/user_input/fastqinfo.tsv (info on both markers)
- asper2/user_input/known_occurrences.tsv (info on both markers)
- asper2/user_input/params.yml (Empty or absent ok)
The snakeconfig.yml looks like this:
project: 'asper2'
subproject: 'run1'
db: 'db.sqlite'
fastqinfo: 'asper2/user_input/fastqinfo.tsv'
fastqdir: 'fastq'
known_occurrences: 'asper2/user_input/known_occurrences.tsv'
params: 'asper2/user_input/params.yml'
blastdbdir: 'vtam_db/COInr_blast_2022_05_06'
blastdbname: 'COInr_blast_2022_05_06'
taxonomy: 'vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv'
Then you compute the ASV tables and the optimization files with default parameters:
snakemake -p --resources db=1 -s snakefile.yml --cores 4 --configfile asper2/user_input/snakeconfig.yml --until asvtable_taxa
snakemake -p --resources db=1 -s snakefile.yml --cores 4 --configfile asper2/user_input/snakeconfig.yml --until optimize
Optimized parameter are specific of each marker.
Therefore, it is simpler to run the optimized filter as in the previous section with two files params_mfzr.yml and params_zfzr.yml for each marker:
- asper2/user_input/params_mfzr.yml
- asper2/user_input/params_zfzr.yml
To run the filter command for each marker, we need to create two readinfo.tsv files for each marker based on asper2/prerun/sorted/readinfo.tsv:
- asper2/user_input/sortedinfo_mfzr.tsv
- asper2/user_input/sortedinfo_zfzr.tsv
Then, we can run the filter and taxassign commands with optimized parameters:
vtam filter --db asper2/db.sqlite --sortedinfo asper2/user_input/sortedinfo_mfzr.tsv --sorteddir asper2/run1/sorted --params asper2/user_input/params_mfzr.yml --asvtable asper2/run1/asvtable_params_mfzr.tsv -v --log asper2/vtam.log
vtam taxassign --db asper2/db.sqlite --asvtable asper2/run1/asvtable_params_mfzr.tsv --output asper2/run1/asvtable_params_taxa_mfzr.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 -v --log asper2/vtam.log
vtam filter --db asper2/db.sqlite --sortedinfo asper2/user_input/sortedinfo_zfzr.tsv --sorteddir asper2/run1/sorted --params asper2/user_input/params_zfzr.yml --asvtable asper2/run1/asvtable_params_zfzr.tsv -v --log asper2/vtam.log
vtam taxassign --db asper2/db.sqlite --asvtable asper2/run1/asvtable_params_zfzr.tsv --output asper2/run1/asvtable_params_taxa_zfzr.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 -v --log asper2/vtam.log
The resulting directory tree looks like this:
asper2
|-- db.sqlite
|-- run1
| |-- asvtable.tsv
| |-- asvtable_params_mfzr.tsv
| |-- asvtable_params_taxa_mfzr.tsv
| |-- asvtable_params_taxa_zfzr.tsv
| |-- asvtable_params_zfzr.tsv
| |-- asvtable_taxa.tsv
| |-- fastainfo.tsv
| |-- merged
| | |-- mfzr_1_fw.fasta
| | |-- ....
| `-- sorted
| |-- run1_MFZR_14ben01_2_mfzr_2_fw_trimmed.fasta
| |-- ...
| |-- sortedinfo.tsv
| |-- ...
|-- user_input
| |-- fastqinfo.tsv
| |-- known_occurrences.tsv
| |-- params.yml
| |-- params_mfzr.yml
| |-- params_zfzr.yml
| |-- snakeconfig.yml
| |-- readinfo_mfzr.tsv
| `-- readinfo_zfzr.tsv
The results of the two markers can be pooled as before:
vtam pool --db asper2/db.sqlite --runmarker asper2/user_input/pool_run_marker.tsv --asvtable asper2/pooled_asvtable_mfzr_zfzr.tsv --log asper2/vtam.log -v
vtam taxassign --db asper2/db.sqlite --asvtable asper2/pooled_asvtable_mfzr_zfzr.tsv --output asper2/pooled_asvtable_mfzr_zfzr_taxa.tsv --taxonomy vtam_db/COInr_vtam_taxonomy_2022_05_06.tsv --blastdbdir vtam_db/COInr_blast_2022_05_06 --blastdbname COInr_blast_2022_05_06 --log asper2/vtam.log -v